Table of Contents
Where this sits. This article is the "why won't spike clear?" node of a larger model: spikeopathy as systemic clearance and tolerance failure under persistent antigenic drive. The RAGE/IL-10 tolerance trap and the mTOR/p53 survival mechanism covered here are the "tolerance corrupts" and "drive persists" arms of that framework. The model, the full lever map, and how the pieces connect live on the Spikeopathy hub.
Spike Persistence (clearance failure)
mean 3.8/5Executive Summary
This document examines the emerging scientific evidence that SARS-CoV-2 and its spike protein can persist in human bodies for months to years after initial infection or vaccination, creating chronic health consequences through multiple interconnected mechanisms that challenge conventional medical understanding of viral clearance.
The central finding is that spike protein persistence is not rare or transient. Multiple independent studies have documented spike protein or S1 subunit persistence in brain tissue, blood monocytes, and cerebrospinal fluid for 15+ months post-infection, with case reports documenting detection up to 709 days post-vaccination. This transforms the conceptual framework from acute viral illness to potential chronic antigen exposure.
The biological mechanisms enabling persistence are multiple and sophisticated:
Cellular senescence pathway: Spike protein triggers TLR7 → p38 MAPK signaling in brain border tissues, creating senescence-like secretory phenotypes that prevent normal cellular clearance and maintain chronic inflammatory states
Immune tolerance mechanisms: RAGE/IL-10 signaling creates protective environments for spike-producing cells, while mTOR/p53 survival pathway dysregulation disables apoptosis and allows spike-loaded cells to evade normal immune surveillance
Lysosomal dysfunction: TMEM106B protein accumulation impairs cellular waste disposal, creating intracellular environments where spike protein accumulates rather than being degraded
The downstream consequences create a multi-system health burden:
Fibrin amyloid microclots ("fibrinaloids") resistant to standard fibrinolytic therapy create microvascular obstruction, explaining the exercise intolerance, brain fog, and dysautonomia that characterize Long COVID syndromes
Herpesvirus reactivation (EBV, HSV-1, HSV-2, CMV) occurs as spike-induced immune dysregulation creates permissive environments for latent viruses, creating overlapping symptom patterns that complicate diagnosis and treatment
Neurovascular stress particularly affects APOE4 genotype carriers, creating specific vulnerability patterns for neurological sequelae including cognitive impairment, dysautonomia, and neurodegenerative processes
The document challenges conventional treatment paradigms by demonstrating that standard anticoagulants fail against amyloid microclots, while evidence-based approaches include:
- Autophagy enhancers (spermidine, time-restricted eating, fasting) to restore cellular cleanup mechanisms
- Nrf2 activators (broccoli sprouts, curcumin) to enhance detoxification pathways
- Oral and gut microbiome support to address systemic inflammation origins
The document ultimately concludes that:
Spike protein persistence is documented in multiple tissue types for durations far exceeding conventional viral clearance timelines, requiring fundamental revision of "post-acute" categorization
Multiple survival pathway hijacks enable spike-loaded cells to evade apoptosis, creating chronic antigen reservoirs that drive ongoing inflammation and immune dysfunction
Downstream effects are systemic including microvascular obstruction, viral reactivation, and neurovascular stress, creating complex multi-system symptom patterns that defy conventional diagnostic categories
Evidence-based terrain support strategies exist that address underlying mechanisms rather than symptom suppression, including autophagy enhancement, Nrf2 activation, and microbiome restoration
Conventional medicine lacks diagnostic tools for detecting amyloid microclots and chronic spike persistence, creating a patient population with organic pathology dismissed as psychosomatic
The broader message is that the "slow burn" model of spike persistence, immune dysregulation, and microvascular dysfunction explains many treatment-resistant syndromes that emerged in the pandemic's wake, and that effective approaches must address underlying mechanisms rather than symptom clusters.
TL;DR
- Spike protein persists in a subset of people for months to years after infection or vaccination (brain, blood, monocytes)
- Mechanisms: Cellular senescence (TLR7 → p38 MAPK), immune tolerance (RAGE/IL-10), lysosomal dysfunction (TMEM106B), mTOR/p53 survival pathway
- Downstream effects: Microclots (fibrinaloids), herpesvirus reactivation (EBV/HSV/CMV), neurovascular stress
- Reality: Not universal, symptom correlation varies, many improve with time
- Practical: Autophagy enhancers (spermidine, fasting), Nrf2 activators (broccoli sprouts), oral/gut health support terrain
If we treated COVID-19 like a two-week respiratory blip, the research now paints a more chronic, multi-system picture. Think "slow burn": antigen persistence, immune perturbations, microvascular changes, and neurovascular stress especially in vulnerable genotypes. Recent discussions highlight emerging evidence on brain-specific persistence.
Part 1: Spike Protein Persistence
Evidence Level: [PR/PP] – Human tissue, autopsy, and cohort studies show persistent spike in subsets CONFIDENCE: HIGH for detection in subsets, MODERATE for clinical relevance
- Autopsy mapping detected viral RNA widely up to ~230 days post-infection. (Chertow et al., Nature 2022)
- In mild cases, a single-centre cohort found tissue RNA/subgenomic RNA weeks–months after infection, and antigen detection associated with long-COVID symptoms. (Zuo et al., Lancet Infect Dis 2024)
- Spike in cells/serum: S1 in non-classical monocytes up to ~15 months after infection (case-control). (Patterson 2022)
- Brain-specific persistence: Human autopsy work reported S1 at the skull–meninges–brain interface. (Rong et al., Cell Host & Microbe 2024; PMID 39615487)
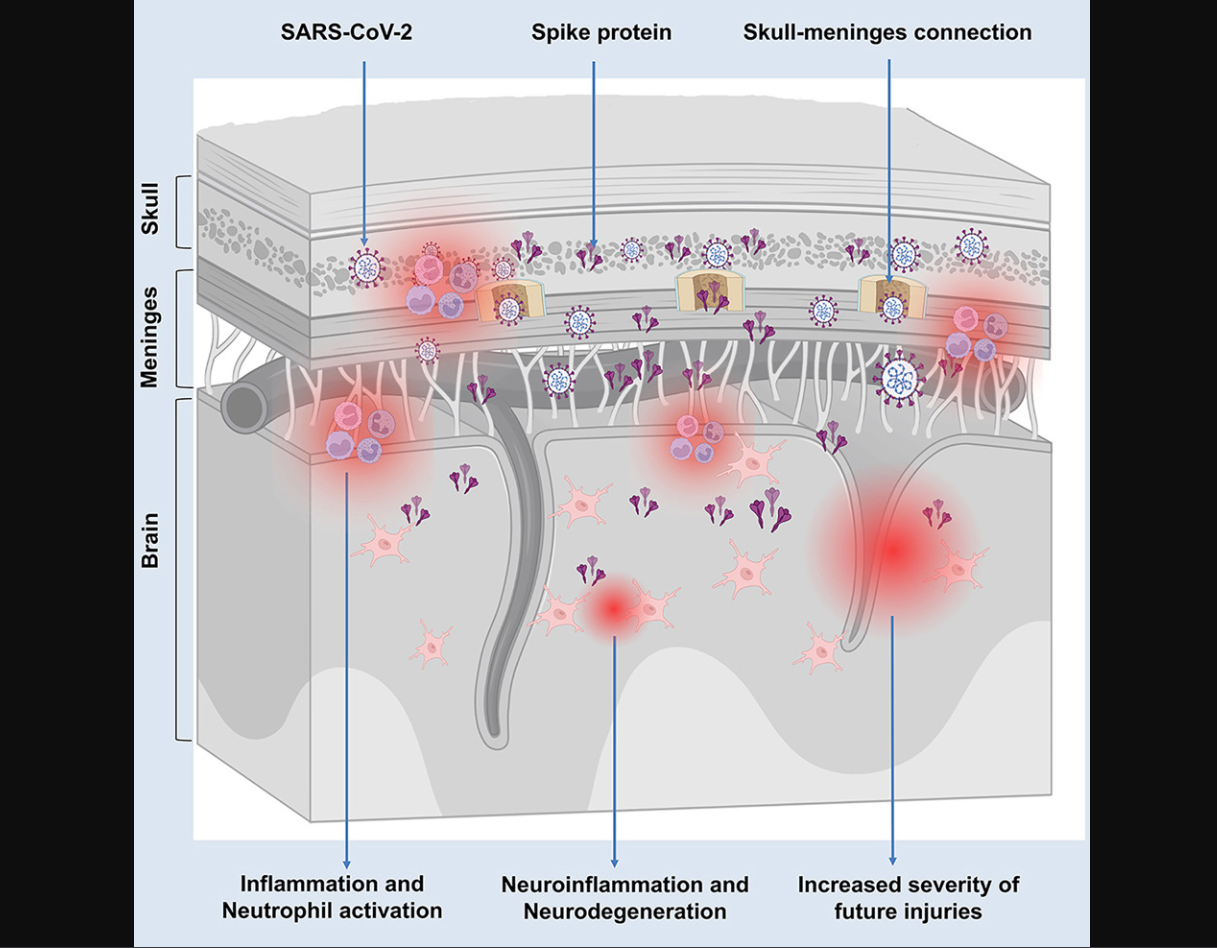
Spike protein S1 persists at the skull–meninges–brain interface post-infection. This anatomical reservoir represents a critical pathway for neuroinflammatory sequelae and may explain neurological Long COVID symptoms. Source: Rong et al., Cell Host & Microbe 2024, Figures 1-2.
Brain endothelial cells & neurovascular invasion: SARS-CoV-2 infection can affect neurological tissues through inflammatory and vascular mechanisms, though direct endothelial spike detection remains an area of active investigation. (Chung et al., 2025, Dis Model Mech, PMID 40195851)
Mechanistic link (2025): A Journal of Neuroinflammation study shows S1 can be taken up into astrocyte endolysosomes and trigger endolysosomal dysfunction and a senescence-like secretory phenotype via TLR7 → p38 MAPK signalling, demonstrating that persistent spike protein alone (even without viral RNA) can provoke ongoing pro-inflammatory signalling at brain border tissues J. Neuroinflammation 2025
PVS preprint: LISTEN reported elevated circulating spike protein in a subset up to 709 days post-vaccination; causality/mechanism unresolved; peer review pending. (medRxiv 2025)
Takeaway: persistence signals exist, but prevalence and clinical meaning differ by cohort and method, and in the context of vaccination, are still being defined in humans. Avoid absolutist language until multi-centre replication matures.
The Cellular Survival Paradox: Why Spike-Producing Cells Don't Die
One of the most critical questions in spike persistence is: Why do cells continue producing spike protein for months when they should have been cleared?
Recent mechanistic insights reveal a troubling answer: spike protein may hijack fundamental cell survival pathways.
HISTORICAL PARALLEL: This persistence playbook isn't unique to spike protein. For over a century, Borrelia burgdorferi (Lyme disease) has used identical survival strategies (round body cysts, biofilm fortresses, intracellular hiding) while the medical establishment pretended persistence was impossible. The pattern repeats: pathogen evolves to survive, medicine evolves to deny it. (See: Neurospirochetosis Evidence)
The mTOR/p53 Survival Mechanism
Analysis by researchers examining persistent spike production proposes that SARS-CoV-2 spike protein activates mTOR (mechanistic target of rapamycin) while simultaneously inhibiting p53, the cell's "guardian of the genome":
- mTOR activation: Promotes cell growth, protein synthesis, and metabolic reprogramming, keeping the cell "alive and productive"
- p53 inhibition: Blocks apoptosis (programmed cell death) and DNA damage responses, preventing the cell from self-destructing
- Net result: Cells that should die continue to survive and produce spike protein
Clinical relevance: This mechanism could explain both viral reservoirs after infection AND prolonged spike production after vaccination, as transfected cells that would normally undergo apoptosis after fulfilling their immunogenic role instead persist.
Supporting evidence:
- Ota et al. (2025) documented spike protein in cerebral arterial walls 17 months post-vaccination in hemorrhagic stroke cases. (PMID: 40184822)
- Patterson et al. (2024) found S1 in monocytes up to 245 days post-vaccination. (medRxiv)
- Video analysis: WeirdSauce interview on mTOR/p53 mechanisms
The Persistence Parallels: Spike Protein vs Borrelia burgdorferi
The survival mechanisms we're documenting in spike protein persistence have disturbing historical precedents. The Lyme disease spirochete has used identical evasion strategies for over a century, while medical authorities denied persistence was possible.
| Persistence Mechanism | Spike Protein (2020+) | Borrelia burgdorferi (100+ years) | Medical Response |
|---|---|---|---|
| Dormant forms | Persister cells, senescence-like state | Round body/cyst forms under stress | Both denied as "impossible" |
| Protected communities | Possible biofilm-like aggregates | Documented biofilm communities (Sapi 2012) | Dismissed as contamination |
| Intracellular hiding | Monocytes, endothelial cells, astrocytes | Fibroblasts, endothelial cells, neural cells | "Antibiotics should reach everywhere" |
| Immune evasion | mTOR/p53 hijack, immune tolerance | Antigenic variation, complement resistance | "Your immune system cleared it" |
| Standard treatment failure | 2-4 week courses don't clear reservoirs | Doxycycline monotherapy fails against cysts | "You must be cured by now" |
THE UNCOMFORTABLE PATTERN: What we call "Long COVID" or "post-vaccine syndrome" follows the exact script used against Lyme patients for decades:
- Deny persistence exists ("it's all cleared")
- Blame the patient ("it's psychosomatic/anxiety")
- Attack dissenting physicians (license threats, guidelines as weapons)
- Protect the narrative at all costs
The Lyme community has been living this nightmare for 40+ years. Their battle proves that persistence is real, denial is systematic, and **healing requires operating outside the failed medical machine.
See full evidence: Neurospirochetosis, Lyme Disease & Multiple Sclerosis
Part 2: Immune Tolerance & Viral Reactivation
Evidence Level: [AN/PR/PP] – Mixed: cell studies for mechanisms, human data for reactivation CONFIDENCE: MODERATE for mechanisms, LOW-MODERATE for clinical significance
- Regulatory/tolerogenic shifts are described in chronic viral states (e.g., HIV Treg/IDO paradigms) and are hypothesized in post-acute COVID contexts. (Jenabian 2014)
- Senescence & SASP as an immune driver: Persistent antigen and chronic innate sensing (now directly shown for S1 in astrocytes via TLR7→p38 MAPK) can induce a senescence-associated secretory phenotype (SASP). SASP factors (IL-6, IL-1β, chemokines) shift local immunity toward chronic innate activation and regulatory feedback loops that can blunt effective adaptive clearance and promote immune drift.
CD169 (SIGLEC-1): A Biomarker for Immune Activation & Spike Persistence
Important distinction: Unlike AXL or efferocytosis pathways which are genuinely hijacked, CD169 is an interferon-stimulated gene (ISG) it increases naturally during viral infections as part of the body's antiviral defense. It is functioning as designed, not being subverted.
What CD169 Reveals About Persistent Spike
Recent research shows CD169+ monocytes persist in Long COVID patients for months after acute infection (Fanelli et al., 2023). This parallels findings of spike protein detection in CD16+ monocytes up to 245 days post-vaccination (Patterson et al., 2024). Why this matters clinically:
| CD169 Expression Pattern | Potential Clinical Meaning |
|---|---|
| Transient elevation (1-2 weeks) | Normal interferon response to infection/vaccination |
| Persistent elevation (>8 weeks) | May indicate active spike reservoir or chronic antigen stimulation |
| High CD169/CD16+ ratio | Could identify patients with spike persistence requiring extended detox protocols |
| CD169+ macrophage expansion in lymph nodes | May explain germinal center persistence (mRNA detected 60+ days) |
Mechanistic insights:
- Interferon-driven marker: CD169 is strongly induced by type I interferons during viral infections (Rincon-Arevalo et al., 2021)
- Inflammation amplifier: CD169+ macrophages, when activated by persistent spike, trigger sustained release of IL-6, TNF-α, and IL-1β (Jalloh et al., 2022)
- Lymph node localization: CD169+ macrophages line lymph node sinuses perfect position for antigen capture and potential mRNA storage
- ACE2-independent entry: CD169 can mediate SARS-CoV-2 entry into macrophages, though infection is typically abortive (doesn't produce new virions) (Jalloh et al., 2022) Clinical utility as a biomarker: CD169 expression could help distinguish between:
- Active spike persistence (requiring extended detox/autophagy protocols)
- Post-viral autoimmune sequelae (requiring immunomodulation)
- Viral reactivation (requiring antivirals)
- Normal immune recovery (requiring only monitoring)
Research gap: Comparative studies of CD169 expression patterns in natural COVID-19 recovery vs post-mRNA vaccination syndromes are needed. Your site is uniquely positioned to document this differential diagnosis framework.
Differential Diagnosis Framework: Long COVID vs Post-Vaccination Syndromes
A critical gap in current research is the lack of biomarker frameworks to distinguish between different post-viral syndromes. CD169 expression, combined with other biomarkers, offers a path toward precision diagnosis.
| Biomarker Pattern | Long COVID (Infection-Induced) | Post-Vaccine Syndrome | Differential Value |
|---|---|---|---|
| Anti-N antibodies | Positive | Negative | Confirms prior infection |
| CD169 expression | Elevated, may persist >6 months | Variable (typically shorter elevation) | Distinguishes active persistence |
| Spike protein (LC-MS) | Detectable in some cases | Detectable in PVS subset | Confirms antigen persistence |
| IL-10/Treg ratio | Often elevated (tolerance trap) | Variable | Immune exhaustion marker |
| EBV/HSV reactivation | Common (30-40%) | Less common | Distinguishes viral reactivation |
| Microclots (Thioflavin T) | Present in ~70% of cases | Present in subset | Microvascular pathology |
| Autoantibodies | Multiple (ANAs, anti-IFN) | Fewer reported | Autoimmune vs toxic |
| Proposed diagnostic algorithm: |
1. Anti-N positive → Prior SARS-CoV-2 infection
├─ Anti-N negative → Vaccine-only exposure
│
2. CD169 + spike protein (LC-MS) assessment
├─ Both elevated → Active spike persistence
├─ CD169 elevated only → Immune activation without persistence
└─ Both normal → Alternative diagnosis
│
3. Viral reactivation panel (EBV, HSV, CMV, VZV)
├─ Positive → Antiviral + immune support
└─ Negative → Continue evaluation
│
4. Autoimmune panel (ANA, anti-IFN, anti-Muscarinic)
├─ Positive → Immunomodulation approach
└─ Negative → Spike-directed detox/autophagy
Why this matters:
- Treatment precision: Patients with active spike persistence may benefit from autophagy inducers (rapamycin, spermidine) and spike-binding agents
- Prognosis: Persistent CD169 elevation correlates with longer recovery times
- Research clarity: Distinguishing subtypes enables more effective clinical trials Call for research: Your site proposes the following research priorities:
- Longitudinal CD169 monitoring in PVS vs Long COVID cohorts
- Correlation of CD169 levels with LC-MS spike protein quantification
- CD169-guided treatment stratification trials
- Standardized flow cytometry protocols for clinical CD169 measurement
TMEM106B: The Missing Lysosomal Link
Your articles document endolysosomal dysfunction (TLR7 → p38 MAPK → senescence in astrocytes), but recent research has identified the specific receptor facilitating this process.
The TMEM106B Discovery
TMEM106B (Transmembrane Protein 106B) is a lysosomal membrane protein that binds spike S1, providing the missing mechanistic link between spike uptake and lysosomal dysfunction:
- Direct binding: Spike S1 binds to TMEM106B's luminal domain
- Endolysosomal accumulation: Facilitates spike concentration in lysosomal compartments
- Lysosomal exocytosis: Enables cell-to-cell transmission without viral replication
- Impaired autophagic flux: Blocks normal lysosomal degradation pathways
Clinical relevance - genetic susceptibility:
TMEM106B genetic variants (particularly the p.T185S risk allele) are associated with:
- Increased susceptibility to frontotemporal dementia (FTD)
- Accelerated neurodegeneration
- Impaired lysosomal function
- Worse outcomes in protein aggregation disorders
Why this matters for spike persistence:
Patients with TMEM106B risk alleles may have:
- Impaired spike clearance from brain tissue and other organs
- Greater susceptibility to long-term neurological sequelae
- Enhanced cell-to-cell spread via lysosomal exocytosis
- Worse neuroinflammatory outcomes due to lysosomal dysfunction
Testing implications:
- TMEM106B genotyping is available through commercial genetic testing services (23andMe, AncestryDNA, etc.)
- Risk allele identification could stratify patients for more aggressive early intervention
- May explain differential neurological outcomes between individuals with similar exposure
Therapeutic considerations:
- TFEB activators (baicalin, trehalose) promote lysosomal biogenesis
- Autophagy inducers may help overcome TMEM106B-related clearance deficits
- Lysosomal pH optimization (niclosamide) may improve degradation capacity
Research priority: Investigation of TMEM106B genotype as a predictor of Long COVID neurological severity and post-vaccination neuroinflammatory outcomes is urgently needed.
Gangliosides: ACE2-Independent Direct Membrane Penetration
Beyond receptor-mediated entry, spike protein can bind directly to gangliosides GM1, GM2, and GM3 sialic acid-containing glycosphingolipids embedded in cell membranes. This provides an ACE2-independent entry mechanism with important implications for neurological involvement.
The Ganglioside Binding Mechanism
What are gangliosides?
- Glycosphingolipids abundant in neuronal membranes
- Key components of lipid rafts (membrane microdomains)
- Concentrated in myelin sheaths and synaptic terminals
- Heavily sialylated (provides negative charge) Spike RBD binding:
- RBD contains a ganglioside-binding domain distinct from ACE2 interface
- Binds to sialic acid residues on GM1, GM2, and GM3 headgroups
- Concentrates spike in lipid rafts for membrane fusion
- Direct membrane penetration without protein receptor requirement
Why this matters clinically:
Tissue Ganglioside Dominance Clinical Implication Brain/neurons GM1 > GD1a > GT1b Explains neurological tropism despite low ACE2 Myelin sheaths High ganglioside density Explains demyelination reports Peripheral nerves GM3, GD1a Explains neuropathy, POTS symptoms Lung epithelium GM3 predominates Contributes to pulmonary infection Immune cells Variable ganglioside profiles Modulates immune cell infection Prion-like connection: Gangliosides play established roles in protein misfolding disorders: - Alzheimer's disease: GM1 promotes Aβ42 aggregation
- Parkinson's disease: α-synuclein binds gangliosides
- Prion diseases: GPI-anchored PrP^C associates with lipid rafts
Spike-ganglioside binding may:
- Accelerate prion-like aggregation of spike itself
- Promote cross-seeding with human prion protein (HuPrP)
- Facilitate amyloid-beta misfolding via GM1 interaction
- Explain neurodegenerative symptom overlap with proteinopathies
ACE2-independent infection explained:
Ganglioside binding resolves a key mystery: how spike infects ACE2-negative cells:
- Olfactory epithelium: Explains anosmia without high ACE2
- Neuronal tissue: Explains neurotropism beyond ACE2 distribution
- Endothelial cells: Explains endothelialitis in low-ACE2 vasculature
- Immune cells: Explains monocyte/macrophage infection mechanisms
Therapeutic implications:
- Ganglioside analogs (e.g., small-molecule competitors) could block binding
- Sialic acid derivatives may act as decoy receptors
- Lipid raft disruptors (statins, omega-3 fatty acids) may reduce infection efficiency
- Membrane stabilizers (sphingolipid precursors) may restore barrier function
Differential vulnerability: Individual variation in ganglioside expression profiles (determined by genetics, diet, and metabolic status) may explain why some patients develop severe neurological manifestations while others do not.
The Immune Tolerance Trap: RAGE, HMGB1, and IL-10
Beyond the TLR7→p38 MAPK pathway, spike protein triggers a parallel tolerance mechanism that may explain why some individuals cannot clear the virus or antigen:
The RAGE/HMGB1/IL-10 Cascade
- RAGE activation: Spike S1 engages RAGE (Receptor for Advanced Glycation End-products) on monocytes and endothelial cells
- HMGB1 release: RAGE activation triggers High Mobility Group Box 1 protein release, a damage-associated molecular pattern (DAMP)
- IL-10 production: The RAGE/HMGB1 axis drives regulatory cytokine IL-10, creating an anti-inflammatory, tolerogenic environment
- ACE2 overexpression: IL-10 paradoxically upregulates ACE2 (the viral receptor). This facilitates viral expansion.
- Blocked clearance: Regulatory T-cell expansion and IL-10 dominance prevent effective viral/antigen elimination
Parallels to HIV tolerance: This mechanism mirrors HIV-associated immune tolerance, where persistent antigen drives IL-10/Treg dominance and progressive immune exhaustion. (PMID: 39615487) Why asymptomatic COVID matters: Some individuals with high IL-10/regulatory responses may remain asymptomatic carriers with ongoing viral replication, a state of "immune ignorance" rather than clearance.
Vaccination-Induced Immune Drift
mRNA vaccination induces similar cytokine profile shifts that parallel early stages of the immune tolerance trap:
- IL-10 elevation alongside IL-6, IL-6R, TNF-α, and IL-1β changes post-vaccination
- Dose-dependent effect: Cytokine alterations markedly higher after 2-3 doses versus single dose
- microRNA dysregulation: miR-21-5p↑ (TLR/NF-κB regulator), miR-23a-3p↓ (NF-κB suppressor), miR-451a↑ (erythroid stress marker)
- Peak timing: 5-6 weeks post-vaccination, overlapping with window for post-vaccine syndrome onset Critically, these molecular changes occurred independently of antibody levels, suggesting vaccination itself can initiate immune drift patterns that mirror early tolerance trap mechanisms (PMC11209245). Clinical relevance: Repeated vaccination may compound pre-existing infection-induced immune dysregulation, potentially accelerating progression toward the tolerance trap state in susceptible individuals.
- Herpesviruses: Meta-analyses/case series document EBV/HSV/CMV reactivation with COVID-19; ICU HSV reactivation has been linked to worse outcomes. (Shafiee 2023; Boers 2024)
- Mechanistic interplay: SASP cytokines, impaired antigen presentation (MHC-I dysregulation reported for ORF8/ORF7a in vitro), and chronic monocyte/endothelial activation create a permissive environment for latent herpesviruses to reactivate, which in turn fuels more inflammation in a feed-forward cycle.
- Transplant/relapse cases underscore risks under profound immunosuppression and show how reactivation can be clinically consequential. (Cureus 2025; PMID 40747163; PMCID PMC12311555) Takeaway: Immune drift in post-acute states likely reflects interacting processes, persistent antigen, senescent cellular secretomes, dysregulated innate sensing and occasional viral reactivation, rather than a single linear cause. This complexity explains heterogeneity in patient presentations and responses to interventions.
Part 3: Microclots & Prion-Like Pathology
Evidence Level: [AN/PR/OBS] – In vitro fibrin studies, observational microclot reports, debated causality CONFIDENCE: MODERATE for mechanism, LOW for clinical impact as primary driver
- In vitro S1 drives amyloid-like fibrin(ogen) resistant to fibrinolysis. (Grobbelaar 2021)
- Long-COVID studies report fibrin amyloid microclots and platelet hyperactivation, with proteomic cargo that could sustain inflammation and hinder microcirculation. (Pretorius/Kruger 2021–2022, open-access review)
- Mechanistic bridge (persistence → microclots): Senescent glia and persistently activated monocytes release SASP cytokines and pro-thrombotic signals (IL-6, tissue factor pathway activators, complement fragments) that promote endothelial activation and platelet hyperreactivity. Endothelial dysfunction plus a fibrin(ogen) structural shift toward amyloid(oid) assemblies creates a milieu where microclots form and persist, reducing effective microvascular oxygen delivery.
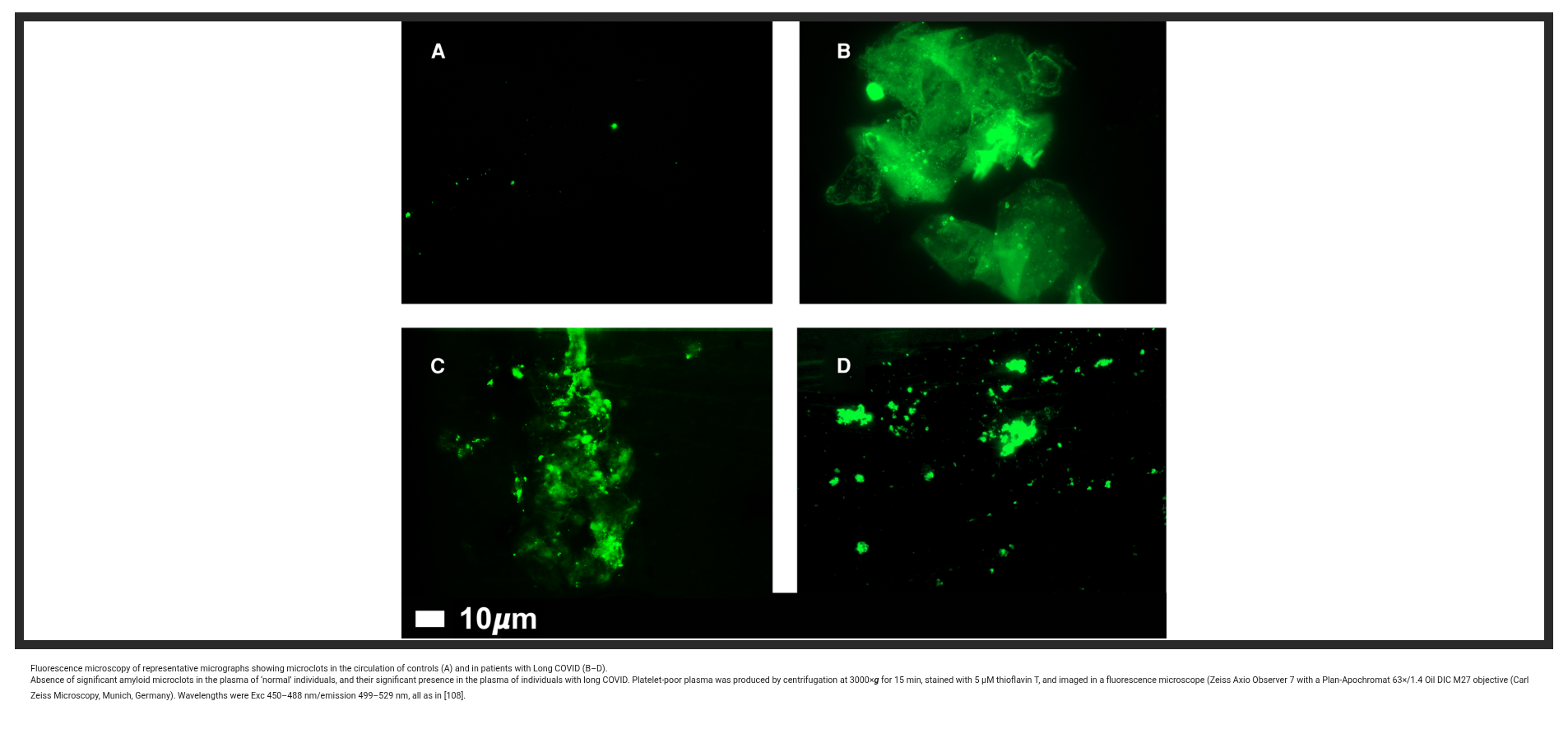
Fluorescence microscopy reveals persistent fibrin amyloid microclots in Long COVID patients. Thioflavin T staining (green) shows resistant fibrin(ogen) aggregates that are resistant to normal fibrinolysis and impair microcirculation. Source: Pretorius & Kell, Biochem J 2022, Figure 4.
Prion-Like Properties: When Spike Becomes a Seeding Template
Beyond its direct effects on fibrin, the S1 subunit itself exhibits troubling prion-like characteristics:
S1 as an Atypical Non-Conventional Prion (ATNC)
Biochemical evidence suggests S1 behaves like a prion, a misfolded protein that templates further misfolding:
- Amyloid fibril formation: S1 forms beta-sheet–rich, fibrillar structures resistant to degradation. (Svensson et al., 2025, ACS Biochem; DOI: 10.1021/acs.biochem.5c00550)
- Accelerated neurodegeneration: Spike S1 accelerates aggregation of human prion protein (HuPrP) and amyloid-beta 42 (Aβ42) in vitro. (Idrees & Sohail, 2023 bioRxiv preprint)
- HIF-1α/p53 synergy: Hypoxia-inducible factor 1-alpha (HIF-1α) interacts with prion protein pathways, potentially linking tissue hypoxia (from microclots) with accelerated protein misfolding. (Jeong et al., 2011; PMID: 22036844)
- ORF6 contribution: SARS-CoV-2 ORF6 protein contains amyloidogenic sequences that may contribute to aggregate formation. (Sprunger & Jackrel, 2023)
- Variant differences in PrLD content: Not all spike variants carry equal prion-like risk. Computational analysis using the PLAAC algorithm identifies Delta (B.1.617.2) as having the highest prion-like domain (PrLD) content and prionogenesis score among major variants, driven by specific RBD mutations. Omicron subvariants show substantially reduced PrLD scores compared to Delta and the original Wuhan strain, potentially translating to lower aggregation propensity. (Tetz & Tetz, 2022, Microorganisms, PMID 35208734) [AN]
- Key RBD residues: Specific residues in the RBD contribute to PrLD characteristics: Q474, N481, Q493, Q498, N501. These positions cluster around the ACE2-binding interface and the heparin-binding site (residues 453-458 and 471-473), suggesting potential for co-aggregation with heparan sulfate and other glycosaminoglycans on cell surfaces. (Same source)
Limitations: These findings are primarily computational predictions and in vitro aggregation assays. Clinical studies comparing prion-like disease outcomes between Delta and Omicron infections are lacking. Whether PrLD differences translate to meaningful differences in long-term pathological aggregation or clinical outcomes remains unknown.
Fibrin–Spike Synergy: The "Calamari Clots"
The interaction between spike and fibrinogen creates hybrid amyloid structures:
- Spike binds fibrinogen directly, inducing conformational changes
- Resulting fibrils display both spike epitopes AND amyloid characteristics
- These aggregates mimic prion "plaques", resistant to normal proteolysis
- Pretorius et al. termed these "calamari clots" for their unusual rubbery, fibrous morphology
Unified framework: The tolerogenic (IL-10), allergenic (RAGE/mast cell activation), and potentially carcinogenic (HIF-1α/p53 dysregulation) aspects of spike persistence may converge via prion-like mechanisms, creating a framework for understanding multi-system, progressive Long COVID symptoms.
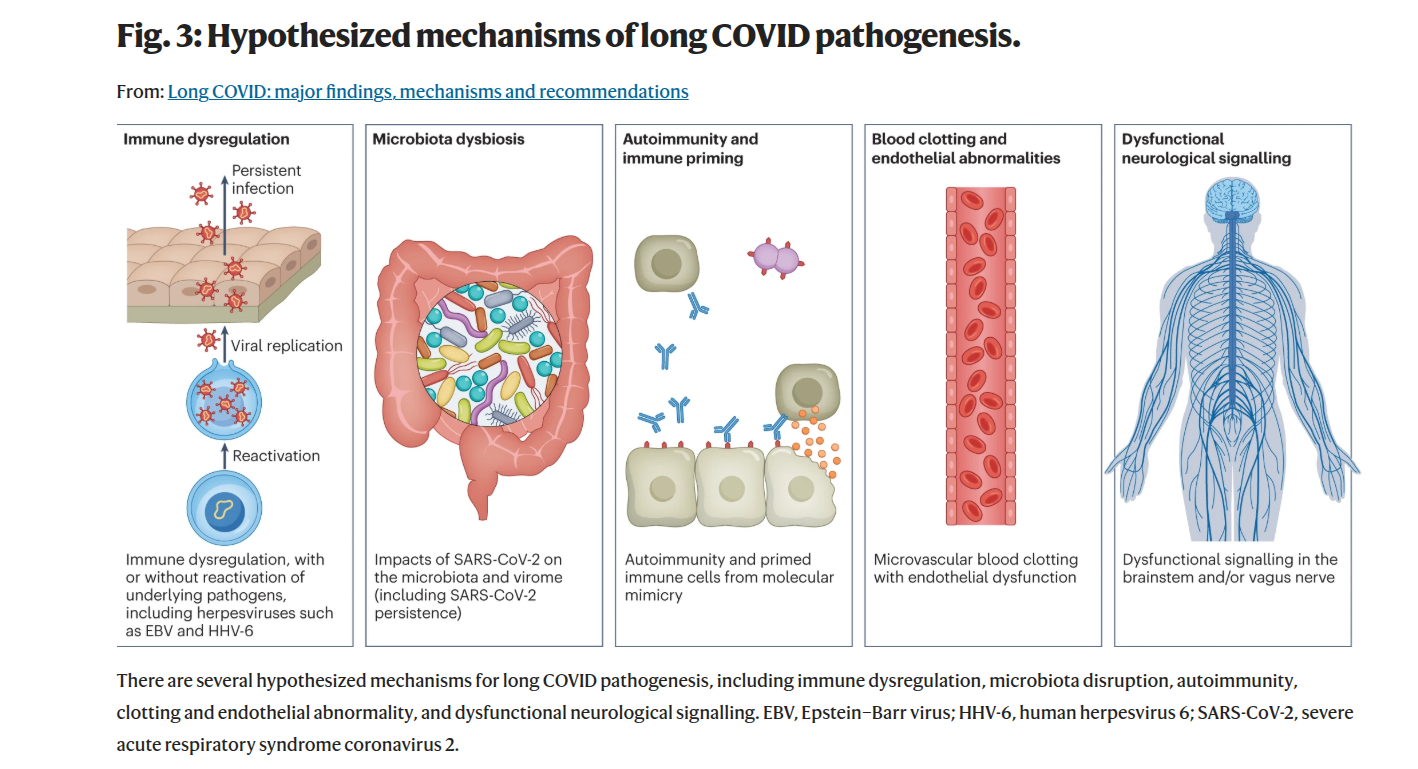
The vicious cycle of Long COVID: persistent spike protein drives chronic immune activation, endothelial dysfunction, and microclot formation, which in turn create a permissive environment for viral reactivation and further antigen persistence. This self-reinforcing loop may explain the progressive and multi-system nature of Long COVID symptoms. Source: Nature Reviews 2023.
- Clinical significance: The presence of microclots correlates with symptoms in several cohorts, but standardisation of assays, causal proof, and interventional data are still required. Small open studies suggest benefit from antiplatelet/antithrombotic strategies in select patients, but robust RCTs are missing.
Takeaway: Persistent antigen → chronic innate signalling → endothelial & platelet activation → prion-like aggregation is a plausible path to microvascular impairment and neurodegeneration; proving causality and safe, effective treatments remains an active research priority.
Part 4: Neurovascular Risk & Barrier Dysfunction
Evidence Level: [AN/CM/PR] – Cell studies for mechanisms, genetic associations for APOE4 CONFIDENCE: MODERATE for vulnerability factors, LOW-MODERATE for population-level impact The breakdown of barrier integrity whether in the gut, oral cavity, or blood-brain barrier is a recurring theme in chronic inflammatory conditions. Research into endotoxemia and systemic inflammation, explored in contexts ranging from sepsis to complex neuroimmune conditions, provides a valuable framework for understanding the multi-system symptoms seen in Long COVID. In this model, a persistent trigger (like spike protein) initiates a cascade of barrier breaches, allowing microbial components (like LPS from the gut or oral cavity) to fuel a chronic, systemic inflammatory state that ultimately converges on the brain's vascular defenses.
- RAGE axis: Multi-omics and cellular work indicate S1–RAGE engagement on monocytes and endothelial cells; the nucleocapsid protein may also interact with RAGE pathways. (Angioni 2023)
- Astrocyte mechanism (2025): A Journal of Neuroinflammation study shows S1 is taken into astrocyte endolysosomes and causes endolysosomal dysfunction that triggers a TLR7 → p38 MAPK cascade, producing a senescence-like phenotype and SASP (IL-6, etc.). This provides a direct cellular route from persistent spike to chronic neuroinflammatory signalling at brain border tissues. (J. Neuroinflammation 2025)
- APOE4 & BBB: APOE4 carriers show hippocampal BBB breakdown independent of Aβ/tau, predicting cognitive decline and vulnerability to peripheral inflammation. (Montagne 2020)
- Peripheral inflammation × APOE4: Systemic LPS or chronic peripheral inflammation exacerbates BBB leak and cognitive deficits in E4/Aβ-prone models, a conceptual parallel to how chronic SASP/monocyte activation could amplify neurovascular injury. (Marottoli 2017)
- Oral–immune link & Metabolic Endotoxemia: This oral–immune link creates metabolic endotoxemia. Oral pathogens like Prevotella intermedia release proteases that degrade CD14 and LBP, critical regulators of endotoxin (LPS). This impairment allows LPS to enter systemic circulation, where it drives chronic inflammation, insulin resistance, and endothelial dysfunction a pathological triad that mirrors key features of Long COVID. (Deschner 2003; Andrukhov 2016)
Synthesis: The mechanistic chain to consider is a multi-barrier cascade: persistent S1 → TLR7 sensing (astrocytes/monocytes) → p38 MAPK activation → endolysosomal dysfunction → senescence/SASP → endothelial & immune activation → BBB stress / microvascular injury. This central nervous system impact is dramatically amplified by concurrent gut/oral barrier dysfunction, which leads to metabolic endotoxemia, creating a state of chronic peripheral inflammation that relentlessly attacks the blood-brain barrier. In APOE4 carriers or people with metabolic dysfunction, this feed-forward loop plausibly accelerates symptomatic decline. Takeaway: Brain-border persistence plus genetic vulnerability and systemic inflammation create intersecting risk nodes for chronic neurovascular and cognitive dysfunction. A Note on Scientific Context: The investigation into immune and barrier dysfunction in chronic conditions has a complex history. In other fields, hypotheses linking endotoxemia and barrier integrity to chronic symptoms have faced significant controversy and required decades of rigorous research to reach consensus. This history supports cautious interpretation of preliminary Long COVID findings and reinforces the value of well-designed clinical trials to avoid premature conclusions.
Practical biomarkers to track BBB integrity & neuroinflammation
Tracking these biomarkers over time provides a quantifiable window into blood–brain barrier integrity, neuroinflammatory tone, and systemic-to-central crosstalk.
How to use: repeat every 8–12 weeks while symptoms evolve; look for directional trends, not single "perfect" numbers.
| Panel | Biomarker | Why it helps (one-liner) |
|---|---|---|
| Core | CRP (hsCRP), fibrinogen | Systemic inflammation & clotting tendency |
| D-dimer | Ongoing fibrin turnover / microthrombi signal | |
| Ferritin, CBC (platelets) | Inflammatory load; platelet activation context | |
| ALT/AST, GGT | Metabolic/ROS stress background | |
| Neuro/BBB | GFAP, sNfL | Astrocytic & axonal injury load over time |
| S100B (if available) | BBB leak proxy in some contexts | |
| TMEM106B genotype | NEW: p.T185S risk allele predicts worse neurodegenerative outcomes & impaired lysosomal spike clearance | |
| Endothelium/Coag | VWF:Ag, ADAMTS13 ratio | Endothelial stress / microangiopathy signal |
| Fibrin-monomer / thrombin time (lab-dependent) | Pro-coagulant milieu | |
| Barrier/Gut–Oral | LPS-binding protein (LBP), sCD14 | Endotoxin exposure/handling |
| Zonulin (caveats) | GI barrier modulation (assay variability) | |
| Metabolic add-ons | fasting insulin, TG/HDL, HbA1c | Insulin resistance & vascular risk context |
| Antigen persistence | Anti-S/Anti-N antibody ratios | Differentiates infection vs vaccination; tracks antigen exposure timeline |
| Spike protein mass spectrometry | Direct detection of circulating spike fragments (research-grade) | |
| Immune tolerance | IL-10 levels | Marks regulatory/tolerogenic shift |
| CD169 (SIGLEC-1) expression | NEW: Interferon-stimulated biomarker distinguishing active spike persistence vs post-viral autoimmunity | |
| Treg frequency (flow cytometry) | Quantifies regulatory T-cell expansion | |
| Prion-like pathology | Thioflavin T fluorescence (blood smear) | Detects amyloid fibrils in circulation (research protocol) |
| Nailfold capillaroscopy | Visualizes fibrinoid deposits in microvasculature |
Clinical decisions belong with your clinician; this is informational context only.
Note on availability: Some advanced markers (spike mass spec, Thioflavin T staining, nailfold capillaroscopy) are research-grade or available only through specialized labs (e.g., Synaptek Labs); they are not standard clinical practice yet. A comprehensive diagnostic framework for spike persistence has been proposed. (Halma & Varon, 2025, Frontiers Med)
Microbiome / Biofilms"] -->|LPS / PGN| LBP["LBP → sCD14
Biomarkers: LBP, sCD14, Zonulin"] LBP --> MONO["Monocytes / Endothelium"] MONO -->|RAGE / TLRs| NFkB["NF-κB / Cytokines
Biomarkers: CRP, Ferritin, CBC"] NFkB --> COAG["Coagulation Shift
Fibrinogen ↑ • D-dimer ↑
Biomarkers: Fibrinogen, D-dimer, VWF/ADAMTS13, Fibrin-monomer"] COAG --> MICRO["Microclots / ↓ O₂ Delivery
Biomarkers: D-dimer"] NFkB --> BBB["BBB Stress
GFAP ↑ • S100B ↑
Biomarkers: GFAP, S100B"] MICRO --> CNS["Neuro Symptoms / Fatigue / Cog
Biomarkers: sNfL"] BBB --> CNS subgraph Tracking Panel CRP["CRP (hsCRP)"] FIB["Fibrinogen"] DD["D-dimer"] GFAPn["GFAP"] SNFL["sNfL"] S100Bv["S100B"] VWFn["VWF:Ag / ADAMTS13"] LBPn["sCD14 / LBP / Zonulin"] Ferritin["Ferritin / CBC"] ALTAST["ALT/AST, GGT"] Metabolic["Fasting Insulin, TG/HDL, HbA1c"] end CRP -.-> NFkB Ferritin -.-> NFkB ALTAST -.-> NFkB FIB -.-> COAG DD -.-> MICRO GFAPn -.-> BBB S100Bv -.-> BBB SNFL -.-> CNS VWFn -.-> COAG LBPn -.-> LBP Metabolic -.-> COAG
Terrain-centric support (adjunctive, not curative), expanded with mechanistic rationale
- Diet quality & polyphenols: Mechanistically these foods can reduce NF-κB signalling and bolster antioxidant defences. Clinical outcome evidence in PASC/PVS is limited. (Review 2024)
- Oral health & biofilms: Treating periodontitis reduces systemic inflammatory burden in other contexts; given the oral→LBP/sCD14 axis, improving oral hygiene may reduce endotoxin load, but individual responses vary.
- Nrf2 activators: Sulforaphane-rich foods plausibly reduce oxidative stress and downstream endothelial activation, mechanistically attractive but unproven for PASC outcomes.
- Sleep, stress, graded activity: Reduce sympathetic drive, cortisol dysregulation and inflammation. Exercise tolerance varies significantly in post-viral states; overexertion can worsen symptoms.
- Mechanistically-informed (experimental) targets: The J. Neuroinflammation TLR7→p38→senescence result points to several hypothesis-generating interventions:
- p38 MAPK modulation: p38 inhibitors have been explored in inflammatory diseases; they are a mechanistic candidate but carry safety and off-target concerns.
- TLR7 pathway dampening: TLR7 antagonism could reduce persistent innate signalling but risks impairing anti-viral defence, clinical trial data are needed.
- Senolytics / senomorphic agents: Removing or modulating senescent cell burden (or reducing SASP) is theoretically attractive to break chronic inflammatory loops; human data in PASC are currently absent and safety must be evaluated.
- Anti-coagulant/antiplatelet strategies: Small, uncontrolled reports suggest symptom benefit in subsets with microclot signals; these approaches require careful clinical oversight and RCT validation.
- Practical medical advice: Any use of experimental or off-label pharmacologic agents must be undertaken by clinicians in the context of informed consent and, where possible, clinical trials.
Disclaimer: These mechanistic targets are hypothesis-generating and not clinical recommendations. Trial evidence is required before routine clinical use.
Therapeutic Implications: Targeting mTOR & Autophagy for Spike Clearance
Given the emerging evidence that spike protein hijacks mTOR signaling to promote cellular survival, researchers have begun exploring whether mTOR inhibitors and autophagy inducers could help clear persistent spike and restore normal cellular function.
The mTOR Connection: Why Targeting This Pathway Matters
The mechanistic target of rapamycin (mTOR) is a serine/threonine protein kinase that regulates:
- Cell growth and proliferation
- Protein synthesis
- Metabolism
- Autophagy, the cell's recycling system
The problem: Spike protein appears to activate mTOR while inhibiting p53, creating a survival advantage for spike-producing cells that should normally undergo apoptosis.
The solution: By inhibiting mTOR, we may:
- Reactivate autophagy, clearing spike protein and damaged cellular components
- Induce apoptosis in persistently infected/transfected cells
- Reduce viral replication (mTOR is hijacked by many viruses)
- Modulate immune responses, reducing chronic inflammation while enhancing antiviral T-cells
mTOR Inhibitors: Research Status
Rapamycin (Sirolimus) and Analogs
What they do:
- Form complex with FKBP12 to inhibit mTORC1
- Promote autophagy by relieving ULK1 inhibition
- Suppress protein synthesis (hindering viral production)
- Enhance stem-like CD8+ T-cells and reduce exhaustion
Evidence in COVID-19 context:
- Preclinical studies: Rapamycin restricts SARS-CoV-2 replication in cell culture
- Kidney transplant patients: On rapamycin showed reduced severity of COVID-19 and lower incidence of pulmonary fibrosis
- Aging resilience: Low-dose rapamycin improves IFN-induced immunity in older adults
- Long COVID hypothesis: By inducing autophagy, could help clear persistent spike remnants
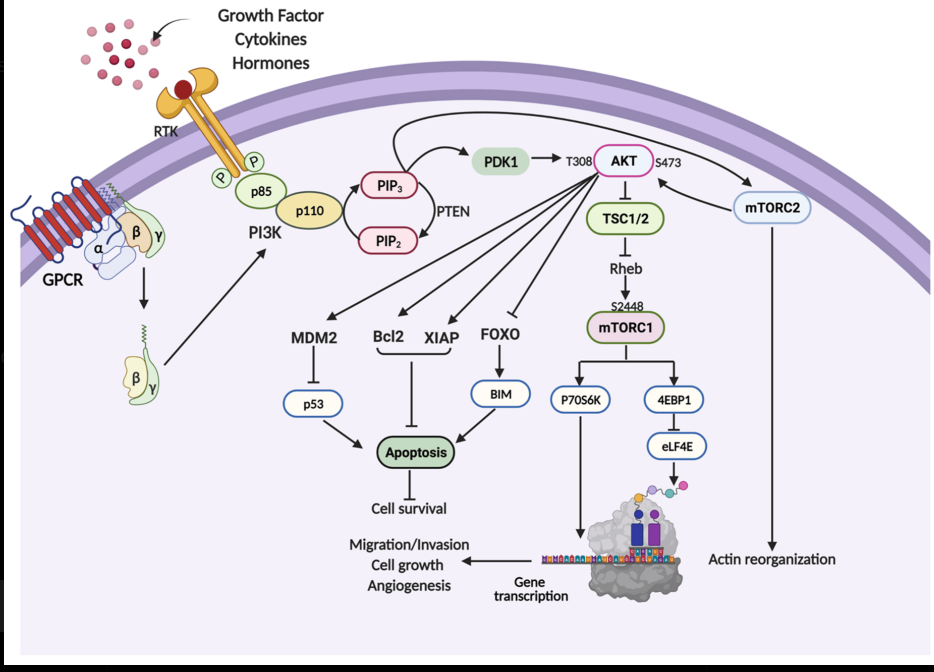
The mTOR pathway integrates signals from nutrients, growth factors, and energy status to regulate cell growth and autophagy. Spike protein hijacks this pathway for persistence.
Safety considerations:
- Can cause mouth sores, hyperglycemia, and immunosuppression
- Timing is crucial, early inhibition may aid viral clearance, but in immunocompromised patients may increase infection risk
- Not available without prescription; clinical trials are required before routine use
Next-Generation mTOR Inhibitors
Drugs like sapanisertib and vistusertib target the kinase domain directly and inhibit both mTORC1 and mTORC2, offering broader effects but with potentially increased side effects. The PI3K-AKT-mTOR axis is hyperactive in many cancers and is hijacked by viruses. Targeting this pathway may have dual benefits in spike persistence and cancer prevention.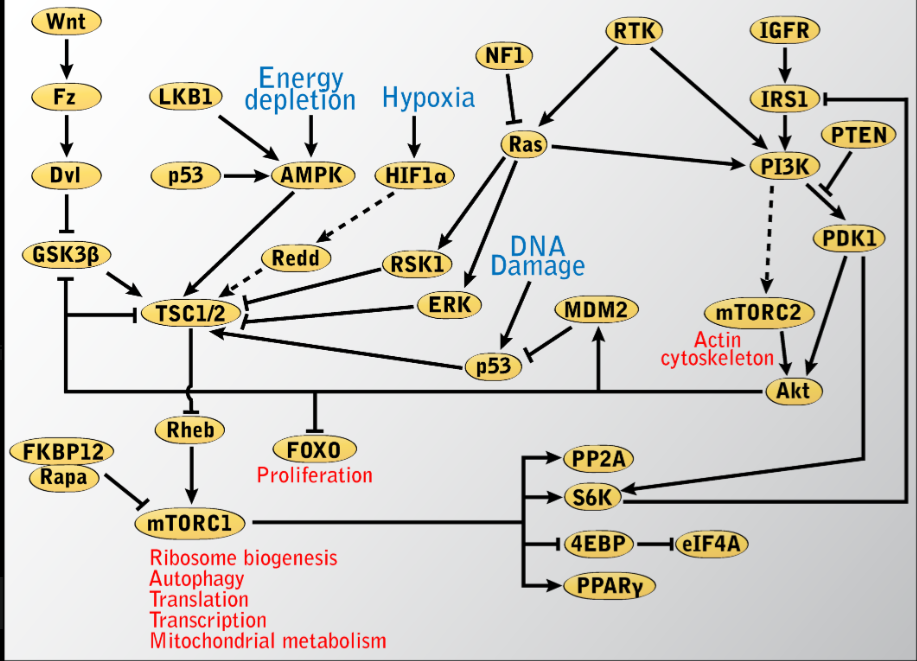
Autophagy: The Body's Cleanup System
What is autophagy? A fundamental cellular process where cells encapsulate damaged components or pathogens in double-membrane vesicles (autophagosomes) that fuse with lysosomes for degradation.
How it helps clear spike:
- Direct degradation (Xenophagy): Autophagy receptors recognize ubiquitinated spike protein and guide it into autophagosomes for breakdown
- Capsid disassembly: During viral entry, autophagy facilitates breakdown of viral proteins
- Immune modulation: Autophagy fine-tunes interferon production and presents viral antigens to T-cells The double-edged sword: Some viruses (including SARS-CoV-2 via ORF3a) can block autophagosome-lysosome fusion, using autophagic structures for replication. This means restoring autophagic flux (not just inducing it) is critical.
Natural Compounds That Induce Autophagy
Several well-studied natural compounds activate autophagy through mTOR-independent pathways:
1. Spermidine
- Found in aged cheese, wheat germ, soy products, mushrooms
- Induces autophagy by inhibiting acetyltransferase EP300
- Shown to extend lifespan in animal models
- Human data: Associated with reduced cardiovascular mortality and cognitive decline
2. Resveratrol
- Activates sirtuin pathways (SIRT1) which promote mitochondrial biogenesis
- Induces autophagy via AMPK activation and mTOR inhibition
- Evidence: Improves endothelial function and reduces oxidative stress in clinical trials
3. Quercetin
- Polyphenol flavonoid found in onions, apples, berries
- Activates autophagy via TFEB (transcription factor EB) nuclear translocation
- COVID-specific data: Small RCTs suggest reduced inflammatory markers
4. Curcumin
- Induces autophagy through multiple pathways including mTOR inhibition
- Evidence: Anti-inflammatory effects documented, but bioavailability is poor
5. EGCG (Epigallocatechin Gallate)
- From green tea
- Activates autophagy via AMPK/mTOR axis
- COVID relevance: Shown to inhibit SARS-CoV-2 main protease in vitro
6. HIF-1α Stabilizers
- Natural compounds that stabilize hypoxia-inducible factor 1-alpha (HIF-1α)
- Have shown efficacy against viruses like Japanese encephalitis by boosting autophagic flux
- Potential application: May help clear spike while supporting cellular adaptation to hypoxia from microclots
Therapeutic Approaches Under Investigation
| Approach | Mechanism | Evidence Status | Notes |
|---|---|---|---|
| Rapamycin/Everolimus | mTORC1 inhibition → autophagy induction | Preclinical + transplant cohort data | Prescription-only; requires monitoring |
| Spermidine supplementation | EP300 inhibition → autophagy | Animal + observational human data | Food supplement with limited long-term safety data in PASC context |
| Nattokinase/Lumbrokinase | Fibrinolytic + may support clearance | In vitro + small pilot studies | May work synergistically with autophagy |
| Double filtration plasmapheresis (DFPA) + SHED-derived secretome | Extracorporeal removal of fibrinaloid microclots, spike protein, immune complexes, and autoantibodies; regenerative support via dental-pulp MSC secretome (IL-10, BDNF, NGF, VEGF, IGF-1) | Established procedural components; no RCT-level outcomes for the combined pathway in Long COVID. McCairn n=38 preliminary cohort data shows significant pre/post anti-spike IgG drop (Wilcoxon p<0.001). Hospital-led programme at Edogawa Hospital, Tokyo. | Full evidence review and author case report (patient 40) in the Amyloid Fibrin Microclots article. Real clinical pathway, not a home intervention; this row is for context only. |
| Polyphenol combinations | Multi-pathway autophagy activation | Small RCTs for inflammation | Bioavailability challenges |
| Intermittent fasting | Natural autophagy induction | Strong animal data, human studies ongoing | Time-restricted eating shows promise |
| NAD+ precursors (NR/NMN) | SIRT activation → autophagy + mitochondrial health | Early human trials for aging | May support energy metabolism in chronic fatigue |
Key Open Questions
- Dosing and timing: What is the optimal window for mTOR inhibition after COVID/vaccination?
- Patient stratification: Who benefits most? (e.g., those with demonstrated spike persistence via LC-MS)
- Combination therapies: Could mTOR inhibitors + autophagy inducers + fibrinolytics work synergistically?
- Biomarker monitoring: How do we track autophagic flux in clinical practice? (LC3-II/I ratios, p62 degradation in research settings)
Clinical Trial field (2025-2026)
As of 2025, several trials are exploring:
- Low-dose rapamycin for Long COVID
- Spermidine-rich diets for post-viral fatigue
- Combination approaches (autophagy inducers + antivirals)
Important caveat: While the mechanistic rationale is strong, clinical outcome data are still pending. Most interventions remain investigational for spike persistence syndromes.
The Verdict
Spike persistence is a real biological phenomenon observed in subsets of people after infection and vaccination. Mechanisms involving cellular senescence, immune tolerance, lysosomal dysfunction, and microclot formation are biologically plausible and supported by in vitro and observational data. If genomic integration occurred, long-term spike production could have significant consequences; this possibility cannot be dismissed.
Reality: True prevalence is unknown: studies vary widely and detection methods differ. Symptom correlation is inconsistent across cohorts. Some people recover without targeted intervention; others remain symptomatic for years. Causality for long COVID as a whole remains unproven. Most evidence sits at the "mechanistically plausible, clinically suggestive" end of the spectrum.
Think of spike persistence as one piece of a larger puzzle: potentially serious for affected individuals, but not a proven explanation for all post-viral symptoms. Terrain supports (autophagy enhancers, Nrf2 activators, oral/gut health) have theoretical rationale but limited clinical evidence and variable safety profiles.
Evidence roundup (curated)
Spike Persistence & Cellular Mechanisms
- Tissue persistence & long-COVID association: Zuo et al., 2024 (Lancet Infect Dis, PMID 38663423)
- Autopsy mapping to 230 days: Chertow et al., 2022 (Nature, PMID 36517603)
- S1 in monocytes up to 15 months: Patterson et al., 2022 (Front Immunol, PMID 35082777)
- S1 in monocytes up to 245 days post-vaccination: Patterson et al., 2024 (medRxiv)
- Skull–meninges–brain spike persistence: Rong et al., 2024 (Cell Host & Microbe, PMID 39615487)
- Spike in cerebral arteries 17 months post-vaccination: Ota et al., 2025 (PMID 40184822)
- Lymph node persistence: Vaccine mRNA and spike protein detected in germinal centers of axillary lymph nodes up to 60 days post-vaccination. (Röltgen et al., 2022, Cell, PMID 35148837)
- Myocardial mRNA: Vaccine mRNA (not translated spike) detected in myocardium within 30 days of vaccination in some autopsy cases. (Krauson et al., 2023, npj Vaccines, PMID 37758751)
- Circulating spike (myocarditis): Free full-length spike protein detected in plasma of adolescents with post-vaccine myocarditis. (Yonker et al., 2023, Circulation, PMID 36597886)
- PVS spike up to 709 days (preprint): Bhattacharjee et al., 2025 (medRxiv)
- PACVS/PACS biomarker framework: Halma & Varon, 2025 (Frontiers Med)
General Persistence & Systemic Impact (Verified)
- Multi-system viral persistence: SARS-CoV-2 may persist in reservoirs for months to years in some patients, with persistent antigen/immune dysregulation as a leading long COVID hypothesis. (Proal et al., 2025, Lancet Infect Dis, PMID 39947217; Peluso & Deeks, 2024, Cell, PMID 39326415)
- Cardiovascular & neurovascular effects: COVID-19 can affect multiple organ systems, including cardiovascular and neurological tissues, through inflammatory and vascular mechanisms. (Chung et al., 2025, Dis Model Mech, PMID 40195851)
CD169 (SIGLEC-1) Biomarker Research (NEW 2025-2026)
- CD169+ monocytes persist in Long COVID: Fanelli et al., 2023 (PMID 38187999)
- CD169 expression correlates with type I interferon responses: Rincon-Arevalo et al., 2021 (PMID 34676541)
- CD169-mediated SARS-CoV-2 infection of macrophages (abortive): Jalloh et al., 2022 (PMID 36279285)
- CD169 as immunoregulatory biomarker in viral infections: Herzog et al., 2022
- High CD169 monocyte:lymphocyte ratio reflects disease severity: Minutolo et al., 2021
- CD169+ macrophages enhance CD8+ T-cell activation: Affandi et al., 2021
TMEM106B & Ganglioside Receptor Research (2024-2026)
- TMEM106B as spike S1 receptor: Baglieri et al., 2023 (PMID 37421949) - Lysosomal membrane protein binds spike, facilitates endolysosomal accumulation and cell-to-cell transmission
- TMEM106B supports Omicron BA.2.86 and JN.1 entry: Saito et al., 2024 (PMID 40556423) - Confirms TMEM106B-mediated viral entry for recent variants
- TMEM106B structural analysis: Nature Immunology, 2023 - Detailed mechanism of ACE2-independent entry via lysosomal pathway
- TMEM106B p.T185S (rs3173615) risk allele: The p.T185 (threonine) variant is the major risk allele associated with increased frontotemporal lobar degeneration with TDP-43 pathology (FTLD-TDP), particularly in GRN mutation carriers. It is linked to higher TMEM106B protein levels and impaired lysosomal function due to slower degradation compared to the protective p.S185 (serine) variant. (Nicholson et al., 2013, J Neurochem, PMID 23742080) TMEM106B functions as a lysosomal transmembrane protein and has been identified as an alternative receptor mediating ACE2-independent SARS-CoV-2 spike binding and viral entry (Baggen et al., 2023, Cell, PMID 37421949). The p.T185S variant affects TMEM106B protein stability and lysosomal localization, which could theoretically influence lysosomal processing or clearance of spike protein; however, this specific connection has not been directly studied.
- Ganglioside binding (GM1/2/3): Sialic acid-containing glycolipids mediate SARS-CoV-2 binding and viral entry (Wang et al., 2021 (PMID 34754101))
- Sialic acid binding bioinformatics: Spike glycoprotein binds host sialic acid glycans (Ahmed et al., 2020 (PMID 32658736))
Immune Tolerance & Viral Reactivation
- HIV-like tolerance mechanisms: Jenabian 2014 (PMC4125509)
- Herpesvirus reactivation (COVID & post-vax reports): Shafiee et al., 2023 (PMID 37559096); Boers et al., 2024 (PMID 39017695)
Microclots & Prion-Like Pathology
- S1-induced amyloid fibrin in vitro: Grobbelaar et al., 2021 (PMID 34328172)
- Long-COVID microclots & proteomics: Pretorius/Kruger 2021–2022 (PMID 34425843)
- Spike-induced amyloid fibrils: Svensson et al., 2025 (ACS Biochem)
- Spike accelerating prion/Aβ42 aggregation: Idrees & Sohail, 2023 (bioRxiv preprint)
- HIF-1α/PrP interactions: Jeong et al., 2011 (PMID 22036844)
- ORF6 amyloidogenic sequences: Sprunger & Jackrel, 2023 (ACS)
RAGE, BBB & Neurovascular Risk
- RAGE pathway & spike S1: Angioni et al., 2023 (Cell Rep Med, PMID 37944530)
- APOE4 → BBB leak & decline: Montagne et al., 2020 (Nature, PMID 32376954)
- LPS worsens E4/Aβ model cognition & BBB: Marottoli et al., 2017 (PMID 28707482)
Oral-Immune & Barrier Dysfunction
- P. intermedia proteases cleave CD14/LBP; sCD14 boosts LPS signaling: Deschner 2003 (PMID 12728301); Andrukhov 2016
Cumulative Damage
- Reinfection risks regardless of vaccination: Bowe et al., 2022 (Nature)
mTOR Inhibitors & Autophagy (NEW 2025-2026)
- mTOR pathway and spike persistence: Melo et al., 2025 (Viruses, PMID 40431629)
- Note: Observational data suggest transplant patients on mTOR inhibitors (sirolimus/everolimus) may have differential COVID-19 outcomes, but results vary by cohort and confounding factors. Clinical trials for rapamycin in Long COVID are ongoing (as of 2025).
- Autophagy in viral clearance: Review 2025
- Spermidine extends lifespan via autophagy: Eisenberg et al., Nature 2016
- Natural autophagy inducers (resveratrol, quercetin, EGCG): Multiple reviews 2020-2025
References and Further Reading
Key Resources
- Nature - Tissue Persistence
- Frontiers - Monocyte Spike
- Lancet - Mild COVID Persistence
- Cell Host Microbe - Brain Spike Persistence
- PNAS Nexus - APOE4 & Prevotella (exploratory association)
- Pretorius Video - Microclots
- X - Nicholas Fabiano Thread on Brain Persistence
- X - Genome Defense
- WeirdSauce - mTOR/p53 in spike persistence
- Frontiers - PACVS/PACS Biomarker Framework
Media & Commentary (clearly non-primary)
These links are useful for context/discussion but are not primary evidence. Keep them separate from references.
- PNAS Nexus, APOE4 & Prevotella (exploratory association)
- Pretorius, Microclots (video overview)
- X, Nicholas Fabiano on brain-border S1
- X, Genome Defense
- X, SARS-CoV-2 spike (S1) can induce cellular aging in human astrocytes
Ethical Declaration (Purpose & Scope)
This article exists to promote scientific transparency, informed consent, and open discussion on biomedical safety. All data are cited from primary or peer-reviewed sources where available. No medical advice is given.
Contested / Active-Debate Notes
- Microclots: Assay methods and clinical utility are evolving; causality vs correlation remains under study.
- PVS spike detection: LISTEN findings are preprint data; interpretation may change post peer review.
- RAGE/S1 and brain persistence: Human autopsy signals exist; translation to population-level risk needs larger, multi-centre studies.
CSI Model: Systems-Level Framework Context
The Comprehensive Spike Interaction (CSI) Model presented in some analyses accurately reflects biologically plausible mechanisms supported by peer-reviewed literature. However, several important caveats apply:
What CSI Gets Right:
- Endothelial ACE2 binding and tight junction disruption mechanisms are well-established
- BBB crossing via adsorptive transcytosis is supported by animal and human data
- CD169+ macrophage involvement in spike dissemination is evidenced
- NF-κB inflammatory cascade and coagulation dysfunction have strong experimental backing
- Microglial activation and synaptic dysfunction align with neuroimaging findings
What CSI Overstates:
- Prevalence: Spike persistence occurs in subsets of patients, not universally. Most people clear spike within weeks; long-term pathological effects at scale remain debated.
- mRNA vs infection equivalence: mRNA vaccines produce transient, localized spike with lower systemic exposure than severe infection. Direct equivalence requires caution.
- Causation certainty: Many effects demonstrated in vitro or in animal models show correlation in human observational studies, but proving they drive chronic disease at scale is ongoing research.
- Prion-like neurodegeneration: Prion-like sequences exist in spike, and protein misfolding hypotheses are mechanistically plausible, but no strong clinical evidence shows increased prion diseases at population levels.
Missing Context:
- Dose/duration dependence (acute high exposure vs. low-level persistence)
- Host factors (genetics, comorbidities, immune status) that explain heterogeneity
- Protective roles of spike-mediated immune activation
- Counter-data showing most vaccinated/infected individuals recover without chronic issues
- Other Long COVID mechanisms (autoimmunity, mitochondrial dysfunction, gut dysbiosis) not directly spike-related
Assessment: The CSI model is a scientifically grounded hypothesis framework that usefully connects dots from existing literature. However, it emphasizes potential chronic harms more definitively than current data warrants. Individual mechanisms have experimental backing, but their population-level clinical impact and interconnections remain areas of active investigation rather than settled science.
Quick FAQ
Is "persistence" the same as ongoing infection? Not necessarily. Studies detect RNA/antigen or protein fragments in tissues/cells; that can reflect residual antigen, low-level replication, or compartmentalized reservoirs. Clinical significance varies by context.
Do vaccines stop brain-border S1 entirely? Mouse data suggest prior vaccination reduces but does not eliminate S1 accumulation/leak at the skull–meninges–brain interface. Are microclots unique to COVID? No fibrin(ogen) amyloid has been reported in other inflammatory states. What's debated is extent, persistence, and impact in Long COVID/PVS.
What's reasonable for self-care? General terrain supports (diet quality, oral health, sleep, graded activity) have varying risk profiles depending on individual health status. Exercise tolerance in particular can worsen symptoms if not carefully paced. Diagnosis/treatment decisions belong with clinicians.
Plain Talk Add-On: The Slow Burn, in real-world language
If science chat makes your eyes glaze over, start here. The short version (30 seconds):
- Pieces of the virus (or its spike) can hang around longer than expected in some people.
- That can keep the immune system slightly "on," so you feel unwell even after "recovering."
- Old viruses like EBV/HSV can "wake up" when your system is stressed.
- Tiny, stubborn microclots can slow oxygen delivery, think thick sludge in skinny pipes.
- Some folks' brain "filter" (BBB) is leakier by genetics (e.g., APOE4), so inflammation hits harder.
A simple analogy:
- Imagine you spilled glitter in your house. You clean, but weeks later, glitter is still turning up in corners.
- Meanwhile, the pipes have a bit of sludge (microclots), so water pressure (oxygen delivery) isn't great.
- If your air filter (brain barrier) is older or thinner, dust gets through more easily. How it can feel day to day:
- Brain fog, short breath on stairs, heavy legs, weird heart flutters, sleep that doesn't refresh, headaches after minor effort, and "good days / crash days" that don't follow a pattern. What this isn't saying:
- Not everyone has this.
- It's not proof of one single cause or a one-size-fits-all cure.
- Research is ongoing; some findings are early or debated.
Practical supports (common-sense, not medical advice):
- Sleep & pacing: Respect your "energy budget" and avoid boom-and-bust cycles.
- Gentle movement: Short walks, light mobility work; increase slowly if tolerated.
- Oral health: Treat gum issues; better mouth health = less background inflammation.
- Food basics: Protein, colorful plants, minimal ultra-processed foods; hydrate.
- Nervous system downshift: Sunlight, breath work, time outdoors, low-stress routines.
- When to get help: New chest pain, severe headaches, one-sided leg swelling, fainting, or stroke-like signs → urgent care. For persistent symptoms, talk to a clinician who takes post-viral issues seriously.
This section is for understanding, not diagnosis. Always work with a clinician for medical decisions.
Microclots & platelet pathology
- Prof. Resia Pretorius, microcirculation & microclots:
- Pretorius & Prof. Doug Kell, latest research and methods:
- Clinical perspective (Laubscher + Pretorius):Brain/BBB context (APOE4)
- Axel Montagne, PhD, BBB dysfunction & APOE4:
- USC Zilkha Seminar (recording link): APOE4 → BBB breakdown
Brain border / persistence
- Spike at skull–meninges–brain interface (paper walk-through)Why: Ties directly to your brain-border section; quick ref recap. EBV / latent virus reactivation (mini-series)
- EBV & Long COVID (Part 1 overview)
- Evidence of EBV reactivation in long haul (study walk-through)BBB & APOE4 (context for vulnerability)
- APOE4 & blood–brain barrier leaks (lay explainer)Neuro / autoimmunity angle
- Brain autoimmunity in Long COVID (MBP, MOG)PVS / LISTEN signals
- Yale LISTEN immune signatures in post-vax syndrome (recap)Clinic-facing overview
- Long-COVID management (broad symptom & systems talk)Clotting/microclots (mechanisms), external link
- Spikeopathies: how spike can drive platelet activation & clotting Watch on React19 →
Acknowledgments
Disclaimer: Informational only; not medical advice.