Table of Contents
TL;DR
Pathologists worldwide are discovering unusual, rubbery "calamari clots" that defy conventional medical understanding. These fibrous masses resist standard anticoagulant treatments and have a unique protein composition.
The emerging picture: Lipid nanoparticles (LNPs) may trigger chronic immune activation through PAD4 enzyme dysregulation, leading to protein citrullination and NETosis—the formation of tough, fibrous clots that blood thinners cannot dissolve.
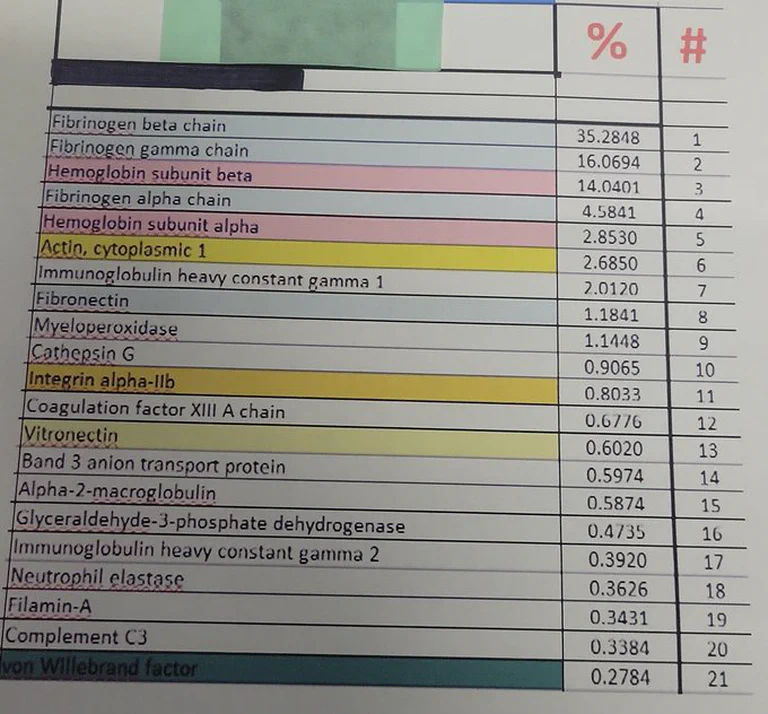
Key evidence: Proteomic analysis reveals fibrinogen dominance (35.28%), hemolysis markers, and NETosis signatures—distinct from normal thrombi.
Why it matters: Conventional anticoagulants fail because they target normal coagulation pathways, not immune-mediated protein corruption.
What Are "Calamari Clots"?
Imagine a blood clot that feels like rubber, resists breakdown, and looks like squid when removed from blood vessels. That's what pathologists are calling "calamari clots"—and they're nothing like normal thrombi.
What makes them different:
- Rubber-like texture that won't dissolve
- Strange protein composition—fibrinogen beta chain at 35.28% (massively overrepresented)
- Don't respond to heparin, warfarin, or other anticoagulants
- Found post-mortem and during surgical procedures worldwide
These aren't classic blood clots. They're immunopathological aggregates—the physical result of your immune system stuck in chronic panic, corrupting the very materials used for clotting.
The Layman's Analogy: A City in Chaos
Think of your body as a well-run city. When a pipe bursts (you get cut), repair crews (platelets) arrive with flexible rubber seals (fibrin) to patch the leak. Once fixed, cleanup crews dissolve the seal. Normal, healthy.
Now imagine what happens with LNPs:
A disruptive delivery truck (the lipid nanoparticle) enters the city, setting off every alarm. The immune system panics. An engineer named PAD4 goes haywire, hardening the rubber seals into brittle plastic. Security guards (neutrophils) throw incredibly sticky webs (NETs) everywhere.
The next "repair" becomes a chaotic clog of hardened plastic, sticky webs, and debris. Standard cleanup crews (blood thinners) have no tools for this mess.
That's the calamari clot in a nutshell.
The Evidence: Proteomic Analysis
Recent protein profiling reveals what's actually in these clots. Think of this as the forensic report from our "city clog."
Fibrinogen dominance with abnormal ratios:
- Fibrinogen β-chain: 35.28% (massively overrepresented)
- Fibrinogen γ-chain: 16.07%
- Fibrinogen α-chain: 4.58% (unusually low)
This skewed ratio indicates disrupted acute-phase response, not normal coagulation. The repair system itself is broken.
Evidence of systemic chaos:
- Hemoglobin subunits (14.04%, 2.85%) — significant hemolysis, damaged red blood cells
- Cytoskeletal proteins: Actin (2.68%), Filamin-A (0.34%) — cellular breakdown
- Immune activation: IgG1 (2.01%), Complement C3 (0.34%)
- NETosis markers: Cathepsin G, neutrophil elastase, myeloperoxidase — the smoking gun
What's missing:
- Factor XIIIa (0.68%) — clot stabilizer, underrepresented
- von Willebrand factor (0.28%) — low for platelet adhesion
Conclusion: These are immunopathological aggregates from cell breakdown, NETosis, and complement activation—not classic thrombi.
The Prime Suspect: Lipid Nanoparticles
The molecular evidence points to LNPs as potential triggers through multiple pathways. Critically, LNPs are not passive carriers—they're biologically active structures that initiate immune and cellular stress responses even without mRNA cargo.
LNP Adduct Formation: Hidden Molecular Damage
LNPs contain ionizable lipids with electrophilic groups that react with amino acids in proteins, creating covalent LNP-protein adducts. These stable bonds:
- Alter protein conformation and enzymatic activity
- Create neoantigens triggering inappropriate immune responses
- Interfere with normal protein-protein interactions
- Modify cellular signaling through structural damage
Preferred targets: Coagulation proteins, endothelial proteins, immune regulators, structural proteins.
The L-DMD Framework (Seger, Gutschi & Seneff)
Groundbreaking research reveals LNPs act as active biointerfaces that disrupt cellular membranes:
- Alter membrane structure, triggering immune responses and ROS
- Interfere with phosphatidylinositol cycle regulating organelle trafficking
- Activate NF-κB, MAPKs, JAK-STAT, mTOR pathways
- Affect PPARγ and cytochrome P450 systems (fat metabolism, detoxification)
- Exosome-mediated spread — LNPs packaged into exosomes for systemic distribution
Biodistribution: Going Everywhere
Visual evidence demonstrates widespread LNP accumulation:
- Multi-organ accumulation occurs rapidly post-injection
- Extended persistence in tissues for weeks to months
- Blood-brain barrier penetration in some formulations
- Reproductive organ accumulation
- Bone marrow deposition
This directly challenges the narrative of "stays at the injection site."
Human Evidence: microRNA Dysregulation
A 2024 pilot study of 111 pregnant women receiving Moderna mRNA-1273 found significant microRNA alterations:
- miR-21-5p↑ — regulates TLR/NF-κB inflammatory signaling, fibrotic pathways
- miR-23a-3p↓ — normally suppresses NF-κB; reduction suggests lost inflammatory brake
- miR-451a↑ — erythroid maturation regulator; suggests hematologic stress
Cytokine changes: IL-6, IL-6R, TNF-α, IL-10, IL-1β peaked 5-6 weeks post-vaccination, markedly higher with 2-3 doses versus single dose.
Critical insight: These changes occurred independently of antibody levels, suggesting LNPs/modRNA themselves drive immune dysregulation.
The Smoking Gun: PAD4 and Citrullination
The enzyme PAD4 (peptidylarginine deiminase 4) emerges as the central mechanism transforming normal coagulation into pathological clot formation.
What PAD4 Does
Citrullination: Calcium-dependent conversion of arginine to citulline. This:
- Alters protein charge, structure, and immune recognition
- Creates rigid, amyloid-like fibrin structures resistant to degradation
The Endotoxin Connection
Geoffrey Pain, PhD and colleagues demonstrated that endotoxin (LPS) is a potent PAD4 activator:
- PAD4 inhibitors protect against LPS-induced lung injury
- C5a/C5aR signaling promotes neutrophil activation and increased PAD4 expression
- Citrullination alters antimicrobial peptides, compromising endotoxin defense
The mechanistic bridge: If bacterial endotoxin robustly activates PAD4, then LNP-induced inflammation likely triggers similar cascades.
Systemic Consequences
Connective tissue implications:
- Collagen citrullination in joints, tendons, bone matrix
- Tissue integrity compromise → tendon fragility, poor wound healing
- Bone demineralization through disrupted collagen cross-linking
PAD4 acts as a "protein reprogrammer" that fundamentally alters structural proteins under inflammatory conditions.
Why Blood Thinners Fail
| Treatment | What It Does | Why It Fails |
|---|---|---|
| Heparins/Warfarin/DOACs | Target coagulation factors to inhibit thrombin | Doesn't calm PAD4, dissolve NETs, or address corrupted platelet surfaces |
| Antiplatelets | Inhibit platelet aggregation | No effect on immune webs (NETs) or citrullinated fibrin |
| Fibrinolytics (tPA) | Dissolves normal fibrin | Struggles with PAD4-modified, amyloid-like structures |
| DNases | Cut NET DNA scaffolds | Experimental, most targeted approach but still unproven |
The failure is loud signal: We're treating immune dysregulation, not simple coagulation problems.
Broader Implications
"Turbo-Cancer" Connection
PAD4 dysregulation creates conditions for aggressive malignancies:
- Chromatin remodeling via histone citrullination
- Tumor suppressor inhibition (p53, ING4, RUNX2)
- NET-mediated DNA damage and mutation promotion
- Immune suppression through T-cell exhaustion
- Angiogenesis and metastasis via NET scaffolding
V-AIDS: Vaccine-Acquired Immune Dysregulation
Progressive T-cell exhaustion manifests as:
- CD8⁺/CD4⁺ depletion and functional impairment
- Viral reactivations (EBV, HSV, VZV, HPV)
- Autoimmune phenomena (Hashimoto's, ANA positivity)
- Senescent cell accumulation
- Mitochondrial dysfunction and energy crisis
A New Path Forward
Given conventional treatment failures, we need multi-targeted approaches addressing root causes (inflammation, PAD4, NETs) rather than just symptoms.
Enzymatic Cleanup
Nattokinase and lumbrokinase — molecular cleanup crews for breaking down tough fibrous structures
Bromelain — complementary circulation support and inflammatory reduction
Caveat: Long-term consequences of breaking down spike protein fragments remain unknown.
Inflammatory Modulation
Curcumin — potent NF-κB inhibition, helps quiet chronic PAD4 activation
Quercetin — calms neutrophil activity, reduces PAD4-mediated damage
Omega-3 fatty acids — membrane stabilization, reduced inflammatory signaling
Resveratrol and OPCs — antioxidant protection against panic-induced oxidative damage
PAD4 Regulation
Vitamin D3 — regulates calcium signaling, keeps PAD4 activity in check
EGCG (green tea) — directly interferes with PAD4 enzymatic activity
Zinc — natural calcium antagonist, additional PAD4 control
Systemic Recovery
Spermidine — enhances autophagy for clearing damaged materials
NMN and NR — restore cellular energy production
CoQ10 and PQQ — stabilize mitochondrial function
Vitamin C — antioxidant support, connective tissue rebuilding
Gentle Support Protocol
For those experiencing post-vaccine syndromes, a structured approach can help restore function while minimizing stress on compromised systems.
Daily Foundation:
- Vitamin D3 (2,000-10,000 IU) + K2 (100-200 μg)
- Magnesium (200-400 mg)
- Omega-3 (1-2 g EPA/DHA)
- Zinc (10-25 mg) with copper (10:1 ratio)
- Vitamin C (250-1,500 mg, liposomal preferred)
Pulse Cycling (6+1 days):
- Mon/Wed/Fri morning: EGCG (200 mg), Quercetin (250 mg)
- Midday: Omega-3, Selenium (50-100 μg), Vitamin C
- Evening: L-glutamine (1.5 g), Magnesium glycinate (300 mg)
- Alternate days: PQQ, CoQ10
- Tue/Thu: Spermidine (1 mg)
- 3-4x weekly: Butyrate (500 mg)
Mini-Senolesis (Week 6, 2 days only):
- Berberine (500 mg) + Turmeric (1 g)
- NAC (250 mg) + Liposomal Glutathione
- Quercetin (500 mg) + Astaxanthin (4 mg)
Contraindications: Avoid during active infection, autoimmune flare, MCAS activation, hypoxia, or adrenal fatigue.
Conclusion
The "calamari clot" phenomenon forces us to reconsider clotting—not as plumbing, but as immune and inflammatory disaster.
These structures are immunopathological hybrids resulting from:
- LNP-induced inflammatory signaling triggering systemic immune activation
- PAD4-mediated protein modification (citrullination)
- Sustained NET formation from dysregulated immune responses
- Endothelial dysfunction from chronic inflammatory damage
The path forward requires:
- Recognition that we're treating immune dysregulation, not just coagulation
- New diagnostics for PAD4 activity and NET formation
- Combination therapies addressing both inflammation and clotting
- Greater caution with technologies that profoundly alter cellular signaling
When the alarm system itself is broken, the solution isn't to silence the repair crews—it's to fix the alarms and calm the panic.
Acknowledgments
This analysis synthesizes research from Nicolina0815 and Narfgb on Substack, whose work on PAD4, citrullination, and LNP-induced immune dysregulation has been instrumental.
Special acknowledgment to Geoffrey Pain, PhD for establishing the endotoxin-PAD4 mechanistic link.
Major acknowledgment to Falko Seger, L. Maria Gutschi, and Stephanie Seneff for the L-DMD framework on LNP-driven membrane dysfunction.
Key References
Primary Research:
- Wang Y, et al. Histone Hypercitrullination Mediates NET Formation — DOI: 10.1083/jcb.200806072
- Lewis HD, et al. PAD4 inhibition disrupts NET formation — PMID: 25622091
- Ndeupen S, et al. mRNA-LNP platform is highly inflammatory — PMID: 34841223
- Zuo Y, et al. NETs in COVID-19 — DOI: 10.1172/jci.insight.138999
Endotoxin-PAD4 Research: 5. PAD4 inhibitor protects against LPS-induced lung injury — PMID: 32889238 6. C5a/C5aR promotes neutrophil PAD4 expression in sepsis — PMID: 39831526
Human Evidence: 7. microRNA dysregulation post-mRNA vaccination — PMID: 38056971 8. Spike persistence in cerebral arteries 17 months post-vaccination — PMID: 40184822 9. DNA contamination in mRNA vials — PMID: 40913499
LNP Biointerface: 10. Seger F, et al. Lipid Nanoparticles as Active Biointerfaces — DOI: 10.20944/preprints202511.0517.v1
Educational content, not medical advice. Clinical decisions belong with qualified healthcare professionals.